O portfólio de Biologia foi sugerido pela professora na sala de aula onde os alunos poderiam pôr assuntos, pesquisas relacionados aos assuntos estudados em sala de aula.
Esse portifólio aborda tais assuntos:
- Sindrómes:
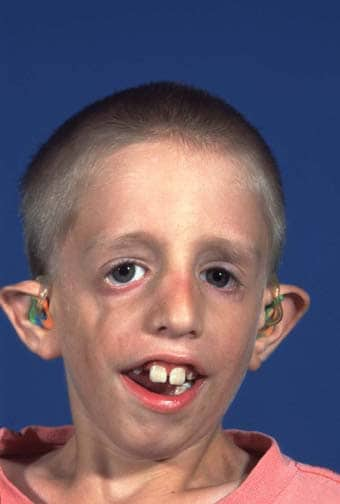
Turner, Klinefelter, Patau, Spoan, Edwards, Workany, Marfan, Cri-Du-Chat, Cornelia de Lange, Treacher Collin, Rett, X Fragil, do XYY, do triplo X.
- Doenças:
Adrenoleucodistrofia, Itabacaninha, Tay-Saches, Parkinson, Alzheimer, Hemofilia, Talassemia, Teste do Pezinho, Hipotiroidismo congênico, Galactossemia , Fenilcetonuria.
-Curiosidades:
Frank Lentini, Medus Van Halen (Pequena miss Sunshine).
No blog foram postados vários assuntos interessantes que devíamos saber, a parte melhor foi a das curiosidades onde foi citado o Frank Lentini, conhecido como o Tripé humano, resumidamente, um cara que nasceu com uma doença, que nele tinham 3 pernas, 2 pênis e 4 pés, eu ainda não tinha visto um caso como esse, não havia conhecido, foi bom parar um pouco pra ler, são curiosidades assim que me interessam.
No blog ainda vocês podem ver como cada doença, síndrome acontece e algumas características, a maioria das síndromes e doenças citadas no blog tem uma foto ou explicando de como acontece ou algum caso acontecido com algum ser humano.
Espero que gostem e que eu possa está ajudando caro leitor.